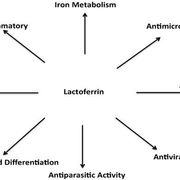
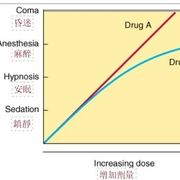
病因學Etiology
病因學Etiology◆因為說了神藥壞話 於是有些人很生氣跑出來大放厥詞:有副作用才有藥效!實是驚為天人感天動地之發現!

造謠生事,對不了解的專業領域大放厥詞後被打臉接著沒招群體攻擊,最後再把議題拋開殭屍般呢喃著高端高端偏離文章主題,實屬高招!
•
藥品效價分析
造謠生事,對不了解的專業領域大放厥詞後被打臉接著沒招群體攻擊,最後再把議題拋開殭屍般呢喃著高端高端偏離文章主題,實屬高招!
•
藥品效價分析也就是俗稱的有沒有效果,有沒有藥效?
◆Pharmacodynamic 藥物靜力學;藥效學
the actions of the drug on the bod 研究藥物對身體產生什麼作用
◆Pharmacokinetic 藥物動力學
the actions of the body on the drug 研究身體對藥物產生什麼作用
◆濃度-作用曲線&致效劑與接受器的結合

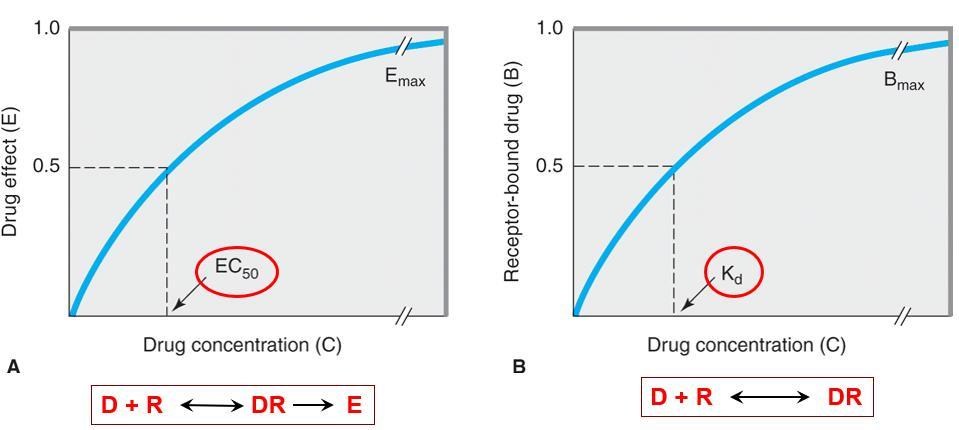
在體外的系統中,藥物濃度(drug concentration)與作用(effect)之間的相關性可依據下面的公式來描述。將濃度(C)以橫坐標,作用(E)以縱座標來表示,劃出濃度與作用的曲線圖,會是一條雙曲線(hyperbolic curve)。
Emax x C
E = -----------------
C + EC50
E: effect 作用;
C: concentration藥物濃度;
Emax: the maximal response that can be produced by the drug 藥物可產生的最大反應;
EC50: the concentration of drug that produces 50% of maximal effect 可以產生50%最大作用的藥物濃度
藥物(D)與接受器(R)結合才能產生作用,藥物濃度增加,結合量(DR)增加,作用增加,但接受器的數目固定,等所有接受器都與藥物結合即達最大作用(Emax),此時若再增加藥物濃度,也無法使作用再增加,因為接受器已被飽和
 藥物劑量及病人的臨床反應
藥物劑量及病人的臨床反應◆要決定用多少劑量的藥物來治療疾病,需先了解藥物的藥效強度(potency)與最大藥效(maximal efficacy)。
藥效強度指的是藥物產生50%最大作用所需的濃度(EC50)或劑量(ED50),取決於藥物與接受器結合的親和力(Kd )及偶合的有效性。
而最大藥效則是藥物所能產生的最大作用(Emax),取決於藥物與接受器交互作用的方式。
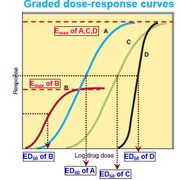
◆在體外系統中,假設A,B,C,D四種藥物均可與相同的接受器結合,產生相同的藥效。但B藥為部分致效劑,因此,B藥的最大藥效(Emax)低於A,C,D藥的最大藥效。

◆而藥物的藥效強度則是取決於藥物與接受器結合的親和力(Kd,以EC50來評估),因此藥效強度的大小是:B>A>C>D,因為只需要很少劑量的B藥即可產生50%的最大作用。
在臨床治療時,藥物的有效性除了要看藥效強度外,也要看最大藥效。B藥雖然藥效強度大於A藥,但A藥的最大藥效大於B藥。因此,雖然服用非常大劑量的B藥,也無法達到A藥的最大藥效。
Median effective dose中間有效劑量(ED50):50% 個體產生療效的劑量
Median toxic dose中間毒性劑量 (TD50) :50% 個體產生毒性的劑量
Median lethal dose 中間致死劑量(LD50) :造成50% 實驗動物死亡的劑量
◆從Quantal dose-effect curves也可得到關於藥物安全閾(margin of safety) 的相關資訊。治療指數(Therapeutic index; TI) : 藥物產生毒性的劑量除以藥物產生療效的劑量,
即TI=TD50/ED50
舉例:
Digoxin:2(使用上需注意,避免引起中毒);Diazepam:1000(使用上較安全)
治療指數小的需要做血中濃度檢測

台版:꧁digoxin 1~2 ng/ml
꧁lidocaine 1~5 μg/ml
꧁gentamicin 5~10 μg/ml
꧁phenytoin 10~20 μg/ml
꧁theophylline 10~20 μg/ml
꧁vancomycin 20~40 μg/ml /中間三苯環以醚(c-o-c)連接/7個amide/含有沒食子酸(pyrogallol)結構
非廣效抑制g(+)厭氧球菌mucopeptide結合在d-AlanylL-d-Alanine末端,形成立體阻礙,使酵素꧁Transglycosidase無法作用,阻止Polymerization
非廣效抑制g(+)厭氧球菌mucopeptide結合在d-AlanylL-d-Alanine末端,形成立體阻礙,使酵素꧁Transglycosidase無法作用,阻止Polymerization
꧁valproic acid 50~100 μg/ml
◆Graded dose-response curve並不適合用於臨床治療時,因為個體之間的差異,相同劑量的藥物在不同病人的反應強度並不盡相同。因此,在臨床上是集合許多實驗動物或病人的反應,劃出Guantal dose-effect curve。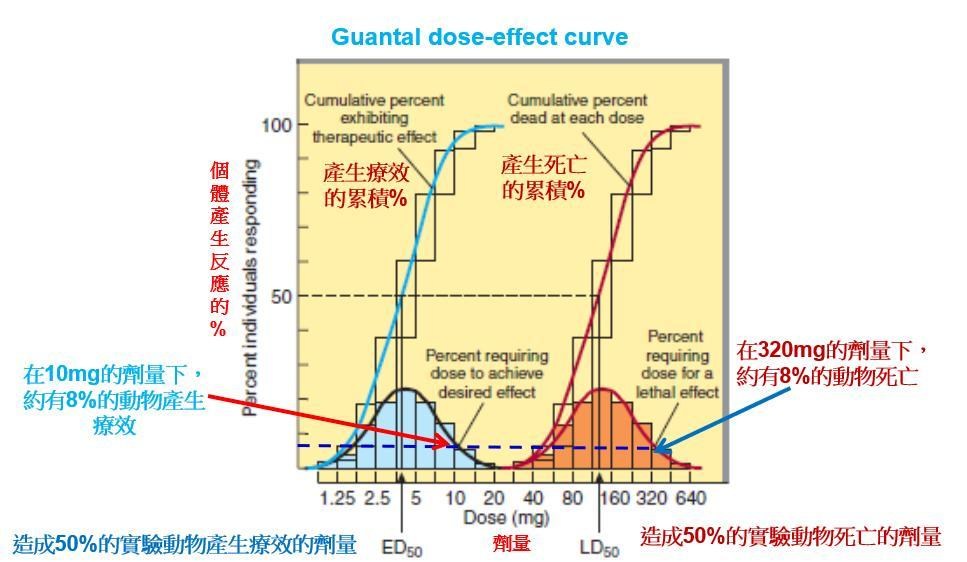
◆所以ED50與TD50根本臨床上是兩個不同的指標,毫無相關!否則要TI何用???
同等劑量下,副作用大小與藥效根本就無關連!否則大家都用動不動就血液惡質,骨髓損傷的癌症化療藥物來治療良性攝護腺腫大好了!保證有效到不行!
那療效毒性的關聯該如何探討呢?
藥物的療效及毒性作用◆在臨床上,一種藥物只會與一種接受器結合,這種藥物的專一性(specific )通常很少見。事實上,一種藥物可能同時會與幾種相似的接受器結合,或是同一種接受器分布在不同的組織,產生不同的生理功能。因此,臨床治療時只能要求藥物的選擇性(selective)夠高,例如對某種接受器或某種組織上的接受器結合親合力較強,而其餘接受器的親和力較差。如此一來,結合親合力強者的生理反應則可視為好處(beneficial effect)或療效(therapeutic effect),而結合親合力弱者的生理作用則可視為毒性作用(toxic effect)或副作用(side effect)
◆藥物的療效及副作用的產生,有三種可能的原因:
1)經由相同的接受器-作用者系統產生療效及毒性:大部分藥物的毒性是其藥理作用(療效)的延伸,如同濃度-作用曲線圖,藥物劑量增加,反應增加,例如降血糖藥可降低糖尿病患者的高血糖,但若劑量太大,降低血糖到低於正常生理濃度,則變成低血糖,可能引起昏迷。
2)經由相同的接受器,但不同的作用者系統或分布在不同組織產生療效及毒性:例如epinephrine的接受器有alpha1,2,beta1,2,3等,又各分布在不同組織,產生不同生理作用,若使用beta2致效劑來緩解急性氣喘症狀,可能也會活化骨骼肌的beta2接受器造成顫震,也會作用在心臟的beta1接受器造成心跳過快。
3)經由不相同的接受器產生療效及毒性:例如三環抗憂鬱藥抑制norepinephrine及serotonin的再回收(reuptake)產生療效,但同時也會拮抗muscrainic接受器產生口乾舌燥、拮抗histamine接受器而想睡覺等副作用。 Ref:Basic & Clinical Pharmacology
藥物動力學藥物動力學的基本參數是清除率(clearance)及分布體積(volume of distribution)。
清除率, 是測量身體將藥物排泄的能力;而分布體積則是測量身體含有藥物的表面空間(apparent space )。
分佈體積(Volume of distribution; Vd) :藥物在身體內的量除以血中濃度
Vd 是一種表面的體積,有時會遠遠超過身體實際的體積。因為Vd的定義是假設身體內為一個均質的溶液狀態,但實際情況是身體內有許多不均質的區室(compartment),例如中樞、胸腔、腹腔、脂肪組織等,而藥物在這些不同區室的分布情況也會有所不同。
但可以從Vd的大小約略得知藥物的分布,若Vd很大,表示血液中藥物的濃度很低,很多藥物都分布到血管外的組織(extra-vascular tissue )。相反地,若Vd很小,則表示藥物大多侷限在血管內,很少分佈到各組織。

清除率(Clearance; CL ):預測的排除速率(rate of elimination)除以藥物濃度(drug concentration; C)。若是腎臟將藥物排除,則腎臟的清除率為CLrenal;若是肝臟將藥物代謝或從膽汁排除,則肝臟的清除率為CLliver;其他器官的排除(CLother)包括肺臟、血液或皮膚等,而全身的清除率(CLsystemic)則是所有器官的清除率加總起來。其中,腎臟及肝臟是排除(elimination)藥物的兩個主要器官。
若藥物是以一級排除(first-order elimination)方式被清除,這時,清除率可以等於劑量(dose)除以曲線下面積(area under the cure; AUC)
CL= dose (mg)/AUC (mg/ml/min) = ml/min
曲線下面積(AUC)是服用一個單一劑量的藥物後,每隔一段時間抽血測血液中的藥物濃度所劃出的曲線圖,橫坐標是時間,縱座標是血中濃度。剛服用藥物後,因藥物被吸收,所以血中濃度增加,曲線往上,但因藥物被排除,且吸收減少,所以曲線逐漸往下。整個曲線下面積即可代表給予一次劑量後,藥物在身體內被排除的速率。

通常少數帶有極性基團或帶電荷的水溶性藥物會從腎臟被排除。但大多數的藥物為親脂性(lipophilic)才容易從腸胃道被吸收,而親脂性的藥物也容易與血漿蛋白結合,與血漿蛋白結合的藥物則無法進入腎絲球,親脂性的藥物若進入腎小管,也很容易被再吸收回血液中,而不容易從腎臟被排除。
此時則需要另外一種排除的方式,即生物轉化作用(biotransformation),也就是代謝(metabolism)。通常代謝是將親脂性的藥物轉化成較極性的親水性代謝物(hydrophilic metabolite),好方便從腎臟排除,而這些親水性的代謝物通常也失去了生物活性。
雖然大多數的藥物的代謝產物失去了生物活性,不具有藥效。但有少數的藥物代謝產物仍具有生物活性,甚至有些代謝產物是具有毒性的。
身體內有些負責代謝藥物等外來物(xenobiotics) 的代謝性酵素,同時也負責合成一些固醇類賀爾蒙(steroid hormones)、膽固醇(cholesterol)、維他命D、膽酸(bile acids)等內生性物質。
由於具脂溶性才容易被吸收,但具水溶性才容易被排除。因此,有些藥物被設計成脂溶性、不具活性的前驅藥(prodrug),被吸收後,才經酵素代謝成有活性的藥物。
大部分的生物轉化作用發生在從腸道吸收進入血液循環之後,到從腎臟排除的過程中,通常是在肝臟。只有非常少數的藥物在腸胃道管腔或管壁內被代謝。所有代謝性的生物轉化作用可被分成兩大類:第一期(phase I )及第二期(phase II )的代謝反應。
Phase I 代謝反應通常是藉由引進或暴露出極性的官能基,例如氫氧基(-OH)、胺基(-NH2)、硫氫基(-SH)等,使藥物代謝物的極性增加。通常這些代謝物會失去活性。少數情況下,代謝物的活性改變或是增強。
許多 phase I 的代謝產物雖然極性增加,但親水性還不夠,還不能馬上從腎臟被排除,通常會再進行phase II的代謝反應。
Phase II的代謝反應是將一些內生性物質,例如葡萄醛酸(glucuronic acid)、硫酸(sulfuric acid)、醋酸(acetic acid)或胺基酸(amino acid) 等水溶性的結合物結合(conjugate)在新形成的極性官能基上,形成高極性的共軛結合物(highly polar conjugate),這些代謝物的結合物即有足夠的親水性從腎臟被排除。
本身是親水性的藥物可不需代謝即直接從腎臟被排除,大多數的藥物通常先進行phase I代謝反應,之後進行phase II代謝反應。但有些藥物本身已具有極性官能基,則直接進行phase II代謝反應。也有些藥物先進行phase II的代謝反應後,其代謝物才進行phase I的代謝反應。
所以

輝瑞連簡單的貯存都有問題了.還談甚麼神藥呢???
以普拿疼為例
在正常治療劑量時,有些藥物的代謝並不會產生毒性,但若超過劑量則會產生有毒的代謝物。
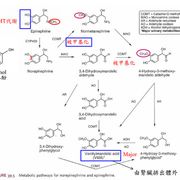
例如普拿疼(acetaminophen)的成人治療劑量為1.2g/d,在此劑量下是相當安全的解熱鎮痛劑。普拿疼本身結構上已有一個-OH的官能基,可直接進行phase II的glucuonidation及sulfation等結合反應(約占95%),phase II的代謝物是沒有毒性的,可直接從尿液排除。
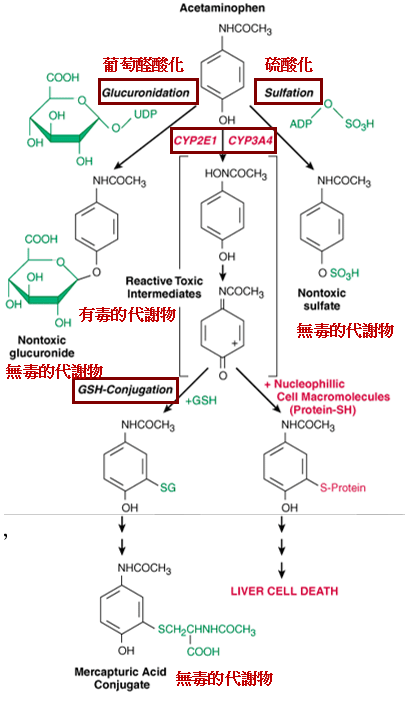
但因內生性物質的量一定,若glucuronidation 及sulfation的路徑被飽和,則其餘少量的普拿疼(約5%)會經由phase I的CYP2E1或CYP3A4代謝成有毒的代謝物,此代謝物很快會與phase II的glutathione (GSH)結合成無毒的代謝物排出。
但若劑量太大,phase II的內生性結合物被用完,又來不及合成新的內生性結合物,則phase I的有毒代謝物則會與細胞內的大分子結合,產生肝毒性(hepatotoxicity)。若在8-12小時內給予N-acetylcysteine,其為GSH的前驅物,可代替GSH與有毒代謝物結合而解毒。